PsychAD Package of Publications
| Key | Title | Authors | Journal | Preprint |
|---|---|---|---|---|
| CAPSTONE | Single-cell atlas of transcriptomic vulnerability across brain disorders | Lee et al |  |
 |
| AGING | Lifespan single-cell transcriptomic atlas of the human prefrontal cortex | Yang et al | |
|
| DISEASE1 | Cell-cell interactome changes across multiple neuropsychiatric and neurodegenerative diseases | Porras et al | TBD | TBD |
| DISEASE2 | Decoding Pathogenic and Protective Gene Regulatory Interactions in Alzheimer’s Disease | Spencer et al | TBD | TBD |
| DISEASE3 | AI-based Characterization of Alzheimer’s Disease Phenotypes from Population Scale Single Cell Data | He et al |  |
|
| DISEASE4 | Personalized Single-cell Transcriptomics Reveals Molecular Diversity in Alzheimer’s Disease | Chandrashekar et al | TBD | |
| GV1 | Single-Nucleus Atlas of Cell-Type Specific Genetic Regulation in the Human Brain | Zeng et al | TBD | |
| GV2 | Single-nucleus transcriptome-wide association study of human brain disorders | Venkatesh et al | TBD | |
| METHOD1 | Fast, flexible analysis of compositional data with crumblr | Hoffman et al | TBD |  |
| METHOD2 | Generalizable Prediction of Alzheimer’s Disease pathologies with Human-Level Accuracy | Ramaswamy et al | TBD | |
| METHOD3 | Efficient differential expression analysis of large-scale single cell transcriptomics data using dreamlet | Hoffman et al |  |
|
| RESOURCE | Population-scale cross-disorder atlas of the human prefrontal cortex at single-cell resolution | Fullard et al |  |
N/A |
CAPSTONE
Single-cell atlas of transcriptomic vulnerability across brain disorders
Donghoon Lee, Mikaela Koutrouli, Nicolas Y. Masse, Gabriel E. Hoffman, Seon Kinrot, Xinyi Wang, Prashant N.M., Milos Pjanic, Tereza Clarence, Fotios Tsetsos, Deepika Mathur, David Burstein, Karen Therrien, Aram Hong, Clara Casey, Zhiping Shao, Marcela Alvia, Stathis Argyriou, Jennifer Monteiro Fortes, Pavel Katsel, Pavan K. Auluck, Lisa L. Barnes, Stefano Marenco, David A. Bennett, PsychAD Consortium, Lars Juhl Jensen, Kiran Girdhar, Georgios Voloudakis, Vahram Haroutunian, Jaroslav Bendl, John F. Fullard, Panos Roussos
doi: https://doi.org/10.1101/2024.10.31.24316513
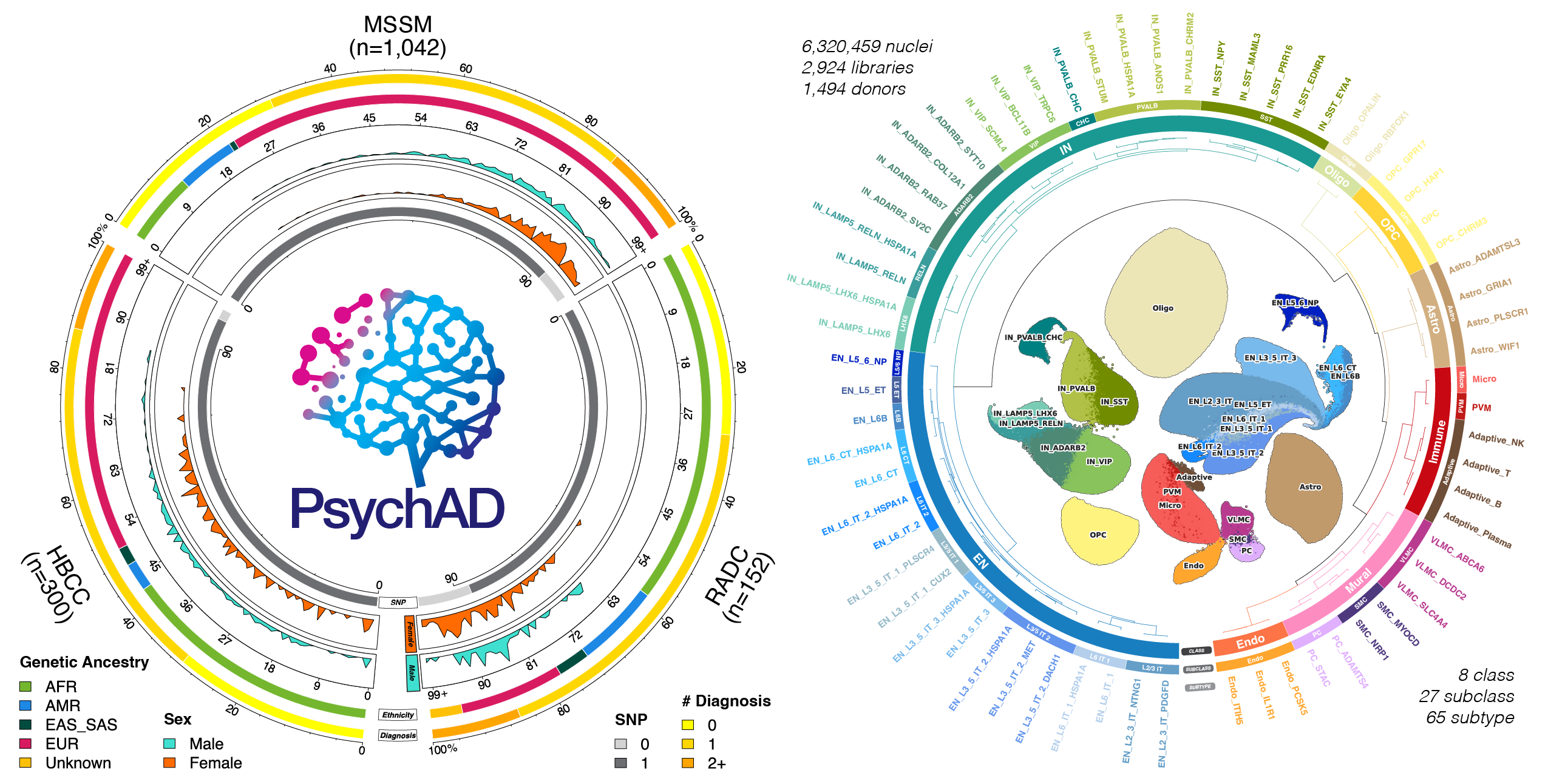
Neurodegenerative and neuropsychiatric diseases impose a significant societal and public health burden. However, our understanding of the molecular mechanisms underlying these highly complex conditions remains limited. To gain deeper insights into the etiology of different brain diseases, we used specimens from 1,494 unique donors to generate a population-scale single-cell transcriptomic atlas of the human dorsolateral prefrontal cortex (DLPFC), comprising over 6.3 million individual nuclei. The cohort includes neurotypical controls as well as donors affected by eight common and complex brain disorders: Alzheimer’s disease (AD), diffuse Lewy body disease (DLBD), vascular dementia (Vas), Parkinson’s disease (PD), tauopathy, frontotemporal dementia, schizophrenia, and bipolar disorder. We show that inter-individual variation accounts for a substantial portion of gene expression variation in the DLPFC. By comparing transcriptomic variation across diseases, we reveal universal signatures enriched in basic cellular functions such as mRNA splicing and protein localization. After discounting these cross-disease signatures, we show strong genetic and transcriptomic concordance among AD, DLBD, Vas, and PD, largely driven by alteration of synaptic signaling functions in neurons. Furthermore, we characterize transcriptomic variation among different AD phenotypes that were distinct from healthy aging. We uncover mitigating effects of interneurons and aggravating effects of immune and vascular cells in AD dementia. Further exploring the effect of the neuropsychiatric symptoms frequently accompanying AD, we identify a link to deep layer excitatory neurons. By constructing transcriptome trajectories that capture AD progression, we show cell-type specific responses implicated in early and late stages of AD. Our atlas provides an unprecedented perspective of the transcriptomic landscape in neurodegenerative and neuropsychiatric diseases, shedding light on shared and distinct processes involving the neuro-immune-vascular systems, and identifying potential targets for therapeutic intervention.
AGING
Lifespan single-cell transcriptomic atlas of the human prefrontal cortex
Hui Yang, Tereza Clarence, Madeline R. Scott, Prashant N.M., Xinyi Wang, Milos Pjanic, Sanan Venkatesh, Aram Hong, Clara Casey, Zhiping Shao, Marcela Alvia, Stathis Argyriou, PsychAD Consortium, Pavan K. Auluck, Stefano Marenco, Vahram Haroutunian, Georgios Voloudakis, Jaroslav Bendl, Colleen A. McClung, Donghoon Lee, John F. Fullard, Gabriel E. Hoffman, Kiran Girdhar, Panos Roussos
doi: https://doi.org/10.1101/2024.11.06.24316592
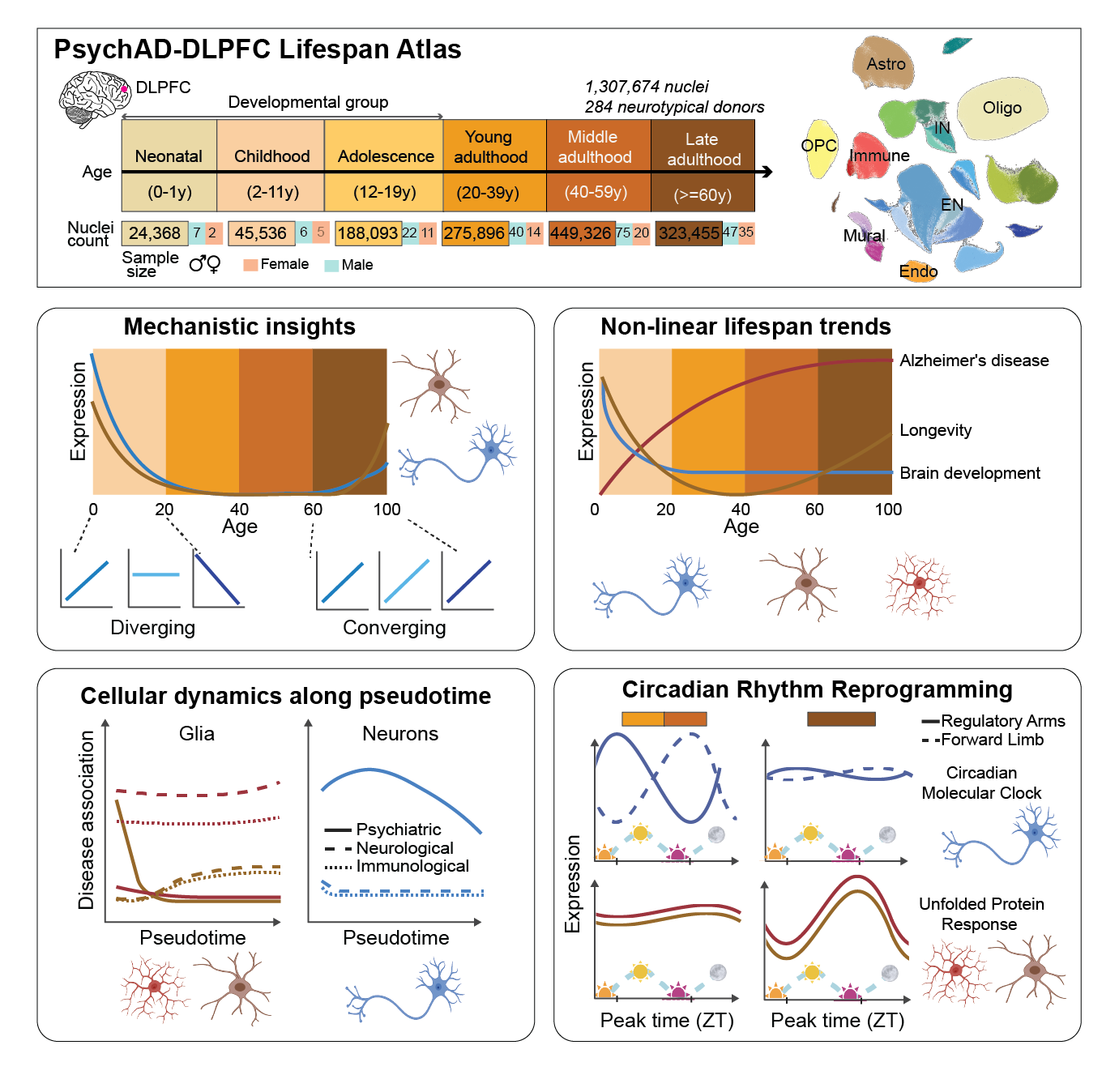
The dorsolateral prefrontal cortex is central to higher cognitive functions and is particularly vulnerable to age-related decline. To advance our understanding of the molecular mechanisms underlying brain development, maturation, and aging, we constructed a detailed single-cell transcriptomic atlas of the human dorsolateral prefrontal cortex, encompassing over 1.3 million nuclei from 284 postmortem samples spanning the full human lifespan (0-97 years). This atlas reveals distinct phases of transcriptomic activity: a dynamic developmental period, stabilization during midlife, and subtle yet coordinated changes in late adulthood. Modeling non-linear age trends across the lifespan shows ten distinct trajectories of the entire transcriptome from all cell types, with notable findings in neurons and microglia, linked to neurodevelopmental disorders and Alzheimer’s disease risk, respectively. Moreover, excitatory neurons exhibit a convergence of gene expression patterns across the lifespan, suggesting the emergence of a common molecular signature of aging. Pseudotime analysis tracing the progression of cellular lineages throughout life reveals key gene clusters with dynamic expression changes that reflect development, maturation, and aging, as well as their connection to brain-related diseases. We uncover significant circadian rhythm reprogramming in late adulthood, characterized by disruption of core clock gene rhythmicity and the emergence of new rhythmic patterns, particularly within microglia and oligodendrocytes. This comprehensive single-cell atlas provides a baseline for understanding the molecular transitions from development through successful aging in the human dorsolateral prefrontal cortex.
DISEASE1
Cell-cell interactome changes across multiple neuropsychiatric and neurodegenerative diseases
Christian Porras, Lyra Sheu, Pramod Bharadwaj Chandrashekar, Roman Kosoy, Milos Pjanic, Jaroslav Bendl, Prashant N.M., Karen Therrien, Deepika Mathur, Steven P. Kleopoulos, Zhiping Shao, Marcela Alvia, Clara Casey, Aram Hong, Stathis Argyriou, David Bennett, George Voloudakis, Vahram Haroutunian, Gabriel E. Hoffman, John F. Fullard, Kiran Girdhar, Daifeng Wang, Donghoon Lee, Panos Roussos
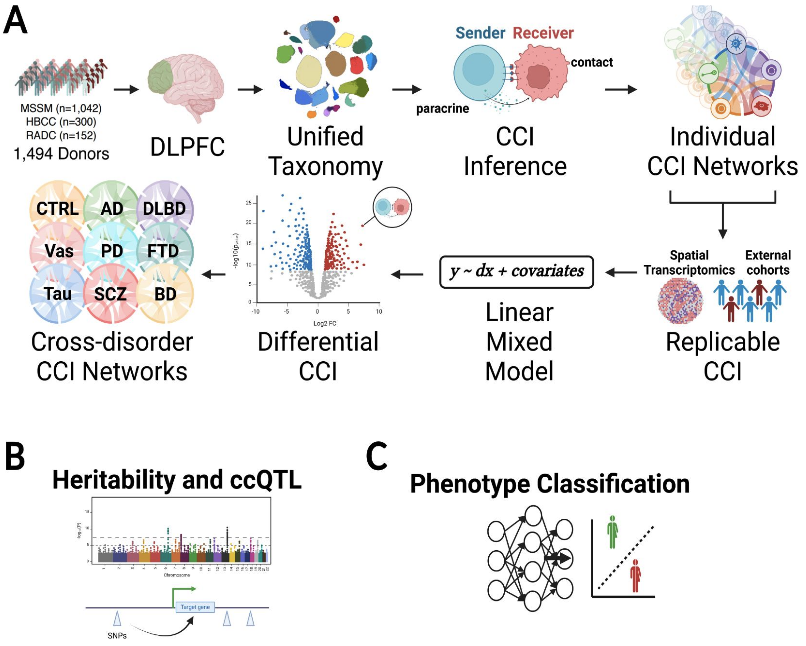
Cell-cell Interactions (CCIs) mediate transcriptomic dysregulation by responding to and altering the microenvironment thereby contributing to the progression of multiple neuropsychiatric (NPD) and neurodegenerative (NDD) diseases, including Alzheimer’s disease (AD), schizophrenia, and bipolar disorder. Recent studies suggest a significant portion of cellular and molecular mechanisms are shared across neurological disorders, despite distinct phenotypic features. Firstly, we focused on generating reproducible CCI networks from a large-scale human brain disease atlas. We inferred over 38 million CCIs between 27 neuronal, glial, and immune cell-types using the PsychAD single-nucleus RNA-seq cohort. We validate these CCIs by ensuring inferred ligand-receptor pairs are colocalized with Visium spatial transcriptomic assays. Secondly, we prioritize 3,063 differentially regulated CCIs mediating eight NPDs and NDDs after accounting for biological and technical variations with linear mixed-effects regression models. We show that during the pathogenesis and progression of NPDs and NDDs, a significant portion of genetic and epigenetic subprocesses are shared and driven by CCIs. We show that about 9% of disease-associated CCIs are shared between NDDs and NPDs, with predominantly immune-glial CCIs differentially dysregulated in NDDs. In contrast, neuronal-glial CCIs are predominantly differentially dysregulated in NPDs. Next, we examined the impact of genetic variation on cell-cell signaling to identify CCI quantitative trait loci (cciQTL). 4429 cciQTLs showed a significant association between CCI scores and the genetic variation in neighboring SNPs of ligand and receptor genes within the CCI. These associations were estimated separately for ligand and receptor genes from 16 sender-receiver cell pairs. A substantial fraction (82.8%) of CCI Genes (cciGenes) showed cell type specificity indicating the presence of unique signaling patterns between specific cell types. Finally, we combine the identified CCIs and whole-genome sequencing into an interpretable deep-learning model to enable personalized NPD and NDD risk prediction from individual genotypes. Our results move beyond independent single-gene analyses, incorporating ligand-receptor gene-pair and cell type-specific interactions to elucidate the higher-order molecular mechanisms underlying NPDs and NDDs, identifying targets for pharmacological intervention.
DISEASE2
Decoding Pathogenic and Protective Gene Regulatory Interactions in Alzheimer’s Disease
Authors TBD
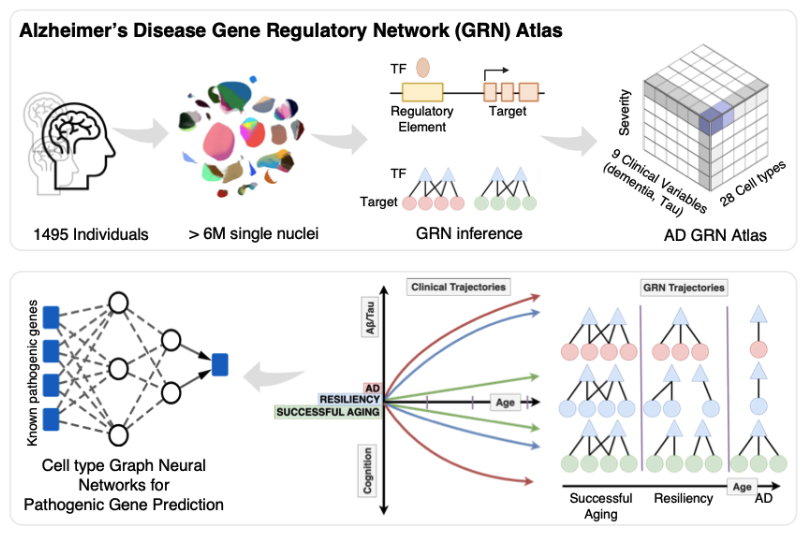
Alzheimer’s Disease (AD) pathogenesis, marked by misfolded protein accumulation and cognitive decline, contrasts with resilience in individuals who maintain cognitive function despite high proteinopathy. Here, we construct a comprehensive AD gene regulatory network (GRN) atlas of the dorsolateral prefrontal cortex across 244 AD, 120 normal aging, and 94 resilient individuals, spanning 27 cell types and 9 clinical measures of AD severity. Using the PsychAD single-nucleus RNA-seq cohort with over 3 million nuclei, we identify 162 transcription factor regulators and prioritize 53 cell type- and disease state-defining regulons by applying a linear mixed model to account for biological and technical variability. Comparative network topology analyses across Braak and CERAD staging (Tau and Aβ burden), age, dementia, and cognitive decline reveal shared and distinct regulatory interactions across AD hallmarks, highlighting key pathogenic transcription factor-target interactions. Next, we build GRNs for AD, control, and resilient groups, performing differential network analysis to identify cell type-specific master regulators that drive AD progression and resilience. Finally, we employ a graph machine learning model to prioritize these master regulators and their target genes. Our findings elucidate cell type-specific gene regulatory networks underlying AD progression and resilience, suggesting potential therapeutic targets for intervention and prevention.
DISEASE3
AI-based Characterization of Alzheimer’s Disease Phenotypes from Population Scale Single Cell Data
Chenfeng He, Athan Z. Li, Kalpana Hanthanan Arachchilage, Chirag Gupta, Xiang Huang, Xinyu Zhao, PsychAD Consortium, Kiran Girdhar, Georgios Voloudakis, Gabriel E. Hoffman, Jaroslav Bendl, John F. Fullard, Donghoon Lee, Panos Roussos, Daifeng Wang
doi: https://doi.org/10.1101/2024.11.01.24316586
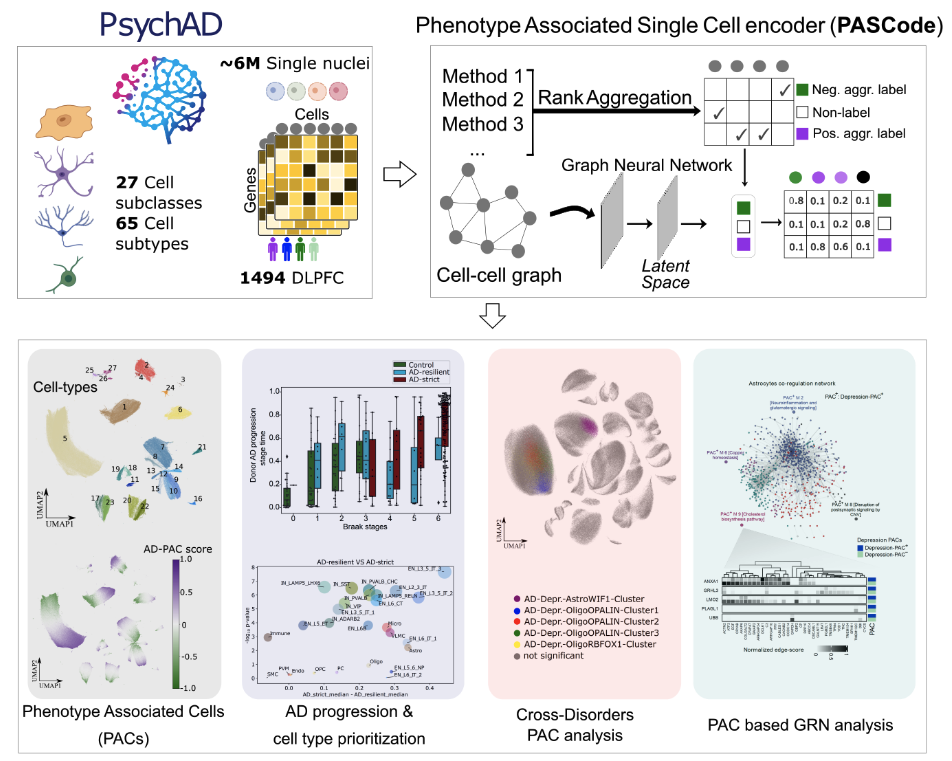
The complexity of Alzheimer’s disease (AD) manifests in diverse clinical phenotypes, including cognitive impairment and neuropsychiatric symptoms (NPSs), yet their etiology remains elusive. Addressing this, we identified phenotype-associated cells (PACs) from 6.3 million nuclei in the prefrontal cortex of 1,494 brains, spanning various AD-related phenotypes. Our deep learning framework, called the Phenotype Associated Single Cell encoder (PASCode), pinpointed ~1.5 million PACs across 27 distinct brain cell subclasses associated with various AD-related phenotypes. Analysis of these PACs revealed the upregulation of an astrocyte subtype with neuroprotective functions in AD-resilient donors. Cross-linking PACs of multiple phenotypes also uncovered a subpopulation of protoplasmic astrocytes that alter their gene expression and regulation in AD donors with depression. Uncovering these cellular and molecular mechanisms underlying the diverse AD phenotypes can offer insights into novel diagnostic markers and therapeutic targets. All identified PACs, along with cell type and gene expression data, are compiled into an AD-phenotypic single-cell atlas for the research community.
DISEASE4
Personalized Single-cell Transcriptomics Reveals Molecular Diversity in Alzheimer’s Disease
Pramod Bharadwaj Chandrashekar, Sayali Anil Alatkar, Noah Cohen Kalafut, Ting Jin, Chirag Gupta, Ryan Burzak, Xiang Huang, Shuang Liu, Athan Z. Li, PsychAD Consortium, Kiran Girdhar, Georgios Voloudakis, Gabriel E. Hoffman, Jaroslav Bendl, John F. Fullard, Donghoon Lee, Panos Roussos, Daifeng Wang
doi: https://doi.org/10.1101/2024.11.01.24316589
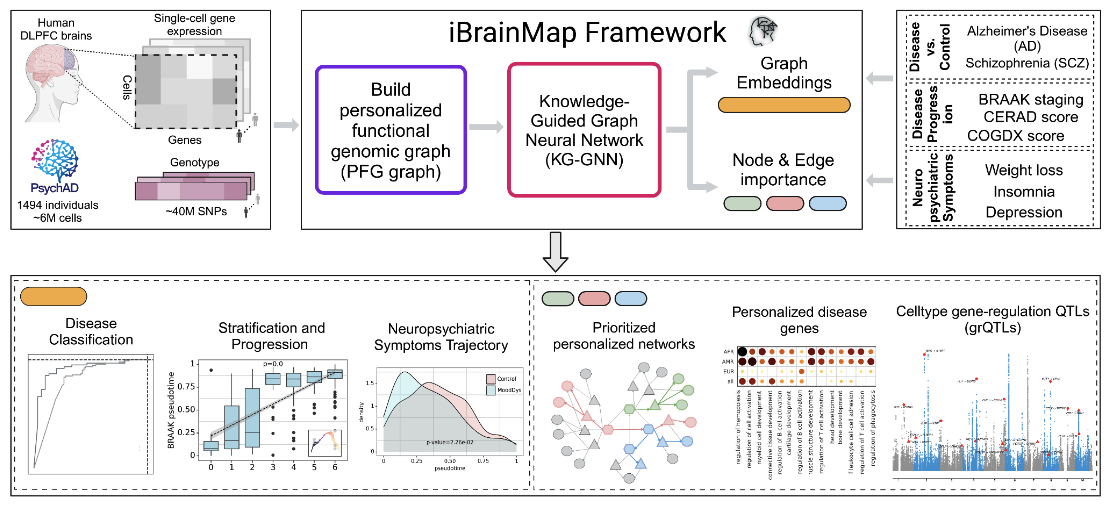
Precision medicine for brain diseases faces many challenges, including understanding the heterogeneity of disease phenotypes. To address this, we present iBrainMap for personalized functional genomics analysis of Alzheimer’s Disease using population snRNA-seq data. Applying iBrainMap to PsychAD data (n=1494), we constructed personalized functional genomic graphs (PFGs) for each individual, incorporating cell type interactions and regulatory information. Using a knowledge-guided graph neural network model, we extracted personalized graph embeddings and importance scores for cell types, genes, and their interactions. Using the graph embeddings, we identified novel AD population subtypes and trajectories. For e.g., population-level AD progression trajectories were closely correlated with plaque pathology and clinical dementia scores. Personalized prioritization of cell types, genes, and their interactions revealed functional genomic subgrouping in disease phenotypes, e.g., identifying ancestry-specific biological pathways. We also identified cell type gene regulatory network changes associated with genetic variants (grQTLs), providing deeper insights into regulatory mechanisms compared to traditional QTL analysis. Our findings are compiled into a comprehensive personalized functional genomic atlas for AD.
GV1
Single-Nucleus Atlas of Cell-Type Specific Genetic Regulation in the Human Brain
Biao Zeng, Hui Yang, Prashant N.M., Sanan Venkatesh, Deepika Mathur, Pavan Auluck, David A. Bennett, Stefano Marenco, Vahram Haroutunian, PsychAD Consortium, Georgios Voloudakis, Donghoon Lee, John F. Fullard, Jaroslav Bendl, Kiran Girdhar, Gabriel E. Hoffman, Panos Roussos
doi: https://doi.org/10.1101/2024.11.02.24316590
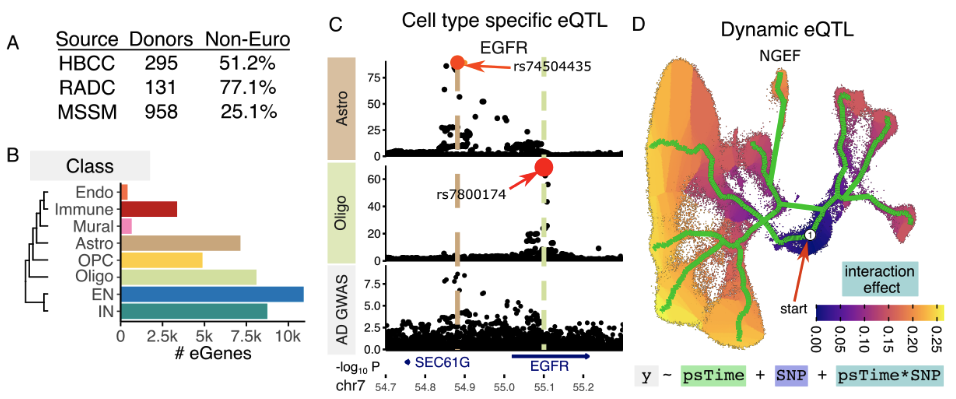
Genetic risk variants for common diseases are predominantly located in non-coding regulatory regions and modulate gene expression. Although bulk tissue studies have elucidated shared mechanisms of regulatory and disease-associated genetics, the cellular specificity of these mechanisms remains largely unexplored. This study presents a comprehensive single-nucleus multi-ancestry atlas of genetic regulation of gene expression in the human prefrontal cortex, comprising 5.6 million nuclei from 1,384 donors of diverse ancestries. Through multi-resolution analyses spanning eight major cell classes and 27 subclasses, we identify genetic regulation for 14,258 genes, with 857 showing cell type-specific regulatory effects at the class level and 981 at the subclass level. Colocalization of genetic variants associated with gene regulation and disease traits uncovers novel cell type-specific genes implicated in Alzheimer’s disease, schizophrenia, and other disorders, which were not detectable in bulk tissue analyses. Analysis of dynamic genetic regulation at the single nucleus level identifies 2,073 genes with regulatory effects that vary across developmental trajectories, inferred from a broad age range of donors. We also uncover 1,655 genes with trans-regulatory effects, revealing distal regulation of gene expression. This high-resolution atlas provides unprecedented insight into the cell type-specific regulatory architecture of the human brain, and offers novel mechanistic targets for understanding the genetic basis of neuropsychiatric and neurodegenerative diseases.
GV2
Single-nucleus transcriptome-wide association study of human brain disorders
Sanan Venkatesh, Zhenyi Wu, Marios Anyfantakis, Christian Dillard, Prashant N.M., David Burstein, Deepika Mathur, Roman Kosoy, Chris Chatzinakos, Bukola Ajanaku, Fotis Tsetsos, Biao Zeng, Aram Hong, Clara Casey, Marcela Alvia, Zhiping Shao, Stathis Argyriou, Karen Therrien, VA Million Veteran Program, PsychAD Consortium, Tim Bigdeli, Pavan Auluck, David A. Bennett, Stefano Marenco, Vahram Haroutunian, Kiran Girdhar, Jaroslav Bendl, Donghoon Lee, John F. Fullard, Gabriel E. Hoffman, Georgios Voloudakis, Panos Roussos
doi: https://doi.org/10.1101/2024.11.04.24316495
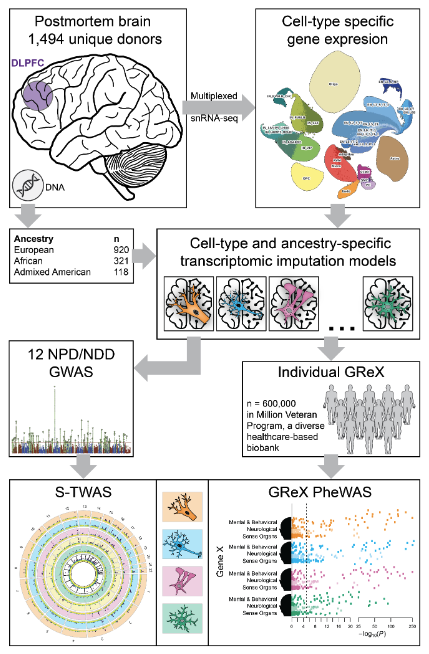
Neuropsychiatric and neurodegenerative disorders exhibit cell-type-specific characteristics, yet most transcriptome-wide association studies have been constrained by the use of homogenate brain tissue, limiting their resolution and power. Here, we present a single-nucleus transcriptome-wide association study (snTWAS) leveraging single-nucleus RNA sequencing of over 6 million nuclei from the dorsolateral prefrontal cortex of 1,494 donors across three ancestries—European, African, and Admixed American. We constructed ancestry-specific single-nucleus-derived transcriptomic imputation models (snTIMs) including up to 27 non-overlapping cellular populations, enhancing the resolution of genetically regulated gene expression (GReX) in the brain and uncovering novel gene-trait associations across 12 neuropsychiatric and neurodegenerative traits. Our snTWAS framework revealed cell-type-specific dysregulation of GReX, identifying over 4,000 novel gene-trait associations not detected by bulk tissue approaches. By applying these snTIMs to the Million Veteran Program, we validated major findings and explored the pleiotropy of cell-type-specific GReX, revealing cross-ancestry concordance and fine-mapping causal genes. This approach enhances the discovery of biologically relevant pathways and gene targets, highlighting the importance of cell-type resolution and ancestry-specific models in understanding the genetic architecture of complex brain disorders.
METHOD1
Fast, flexible analysis of compositional data with crumblr
Gabriel E. Hoffman, Panos Roussos
doi: https://doi.org/10.1101/2025.01.29.635498
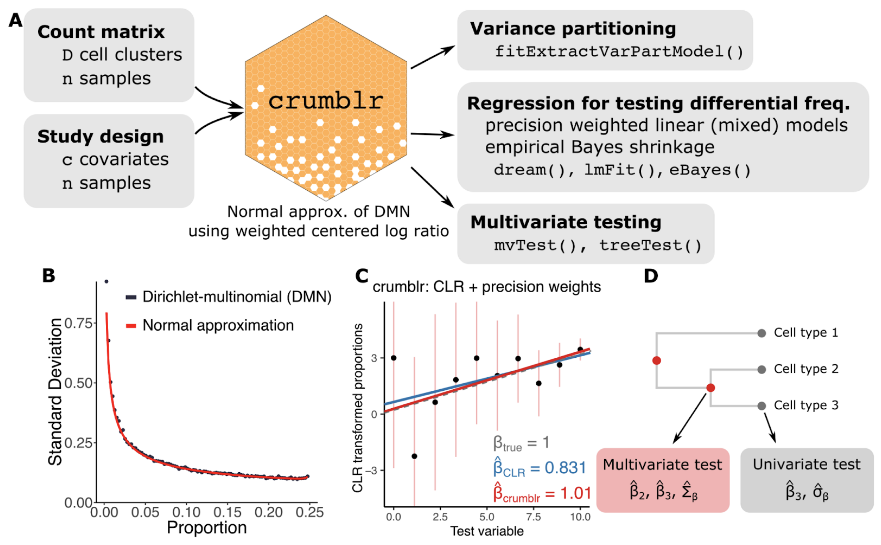
Changes in cell type composition play an important role in health and disease. Recent advances in single-cell technology have enabled measurement of cell type composition at increasing cell lineage resolution across large cohorts of individuals. Yet this raises new challenges for statistical analysis of these compositional data to identify changes associated with a phenotype. We introduce crumblr (diseaseneurogenomics.github.io/crumblr), a scalable statistical method for analyzing count ratio data using precision-weighted linear models incorporating random effects for complex study designs. Uniquely, crumblr performs tests of association at multiple levels of the cell lineage hierarchy using multivariate regression to increase power over tests of a single cell component. In simulations, crumblr increases power compared to existing methods, while controlling the false positive rate. We demonstrate the application of crumblr to published single-cell RNA-seq datasets for tuberculosis infection in T-cells, SARS-CoV-2 infection in blood, and aging of the human brain.
METHOD2
Generalizable Prediction of Alzheimer’s Disease pathologies with Human-Level Accuracy
Vivek Gopal Ramaswamy, Monika Ahirwar, Genadi Ryan, Brittany Dugger, Osama Al Dalahmah, Maxim Signaevsky, Vahram Haroutunian, Steven Finkbeiner
doi: https://doi.org/10.1101/2023.05.19.541376

Characterizing neuropathology in Alzheimer’s disease (AD) is laborious and time consuming, and susceptible to intra- and inter-observer variability. The lack of high throughput approaches to reliably assess neuropathology hampers efforts to use pathology as a means to link clinical features of AD to molecular pathogenesis in the ever growing datasets of AD patients. To remove this roadblock, we designed a computational pipeline that analyzes postmortem tissue from AD patients in a fully automated and unbiased manner in only 20 minutes per whole slide image, a fraction of the time taken with conventional approaches. We trained algorithms to detect, classify, and segment different types of amyloid pathology with a Mask Regional-Convolutional Neural Network. To establish ground truth for training and validation, we developed a tool that collects consensus annotations of neuropathology experts. Our algorithms accurately identified amyloid pathology in samples unrelated to the training dataset, indicating that they detect generalizable pathology features. Our design makes it possible to reconstruct a map of pathology across the entire whole slide image, facilitating neuropathological analyses at multiple scales. Quantitative measurements of amyloid pathology are correlated with the severity of AD measured by standard approaches. Our computational pipeline should enable rapid, unbiased, inexpensive, quantitative and comprehensive neuropathological analysis of large tissue collections and integration with orthogonal clinical and multi omic measurements.
METHOD3
Efficient differential expression analysis of large-scale single cell transcriptomics data using dreamlet
Gabriel E. Hoffman, Donghoon Lee, Jaroslav Bendl, Prashant N.M., Aram Hong, Clara Casey, Marcela Alvia, Zhiping Shao, Stathis Argyriou, Karen Therrien, Sanan Venkatesh, Georgios Voloudakis, Vahram Haroutunian, John F. Fullard, Panos Roussos
doi: https://doi.org/10.1101/2023.03.17.533005
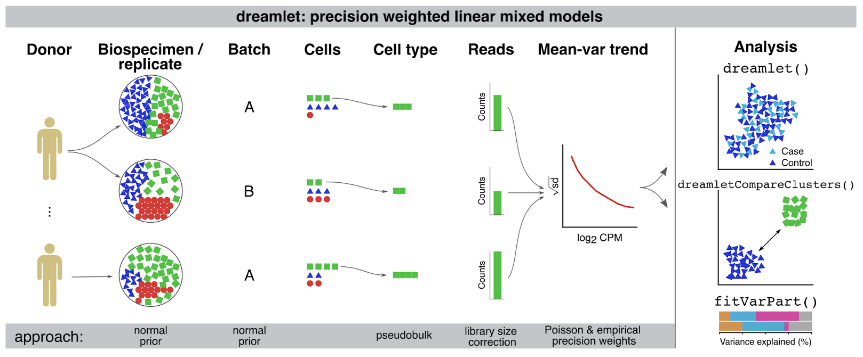
Advances in single-cell and -nucleus transcriptomics have enabled generation of increasingly large-scale datasets from hundreds of subjects and millions of cells. These studies promise to give unprecedented insight into the cell type specific biology of human disease. Yet performing differential expression analyses across subjects remains difficult due to challenges in statistical modeling of these complex studies and scaling analyses to large datasets. Our open-source R package dreamlet (DiseaseNeurogenomics.github.io/dreamlet) uses a pseudobulk approach based on precision-weighted linear mixed models to identify genes differentially expressed with traits across subjects for each cell cluster. Designed for data from large cohorts, dreamlet is substantially faster and uses less memory than existing workflows, while supporting complex statistical models and controlling the false positive rate. We demonstrate computational and statistical performance on published datasets, and a novel dataset of 1.4M single nuclei from postmortem brains of 150 Alzheimer’s disease cases and 149 controls.
RESOURCE
Population-scale cross-disorder atlas of the human prefrontal cortex at single-cell resolution
John F. Fullard, Prashant N.M., Donghoon Lee, Deepika Mathur, Karen Therrien, Aram Hong, Clara Casey, Zhiping Shao, Marcela Alvia, Stathis Argyriou, Tereza Clarence, David Burstein, Sanan Venkatesh, Pavan K. Auluck, Lisa L. Barnes, David A. Bennett, Stefano Marenco, PsychAD Consortium, Kiran Girdhar, Vahram Haroutunian, Gabriel E. Hoffman, Georgios Voloudakis, Jaroslav Bendl, Panos Roussos
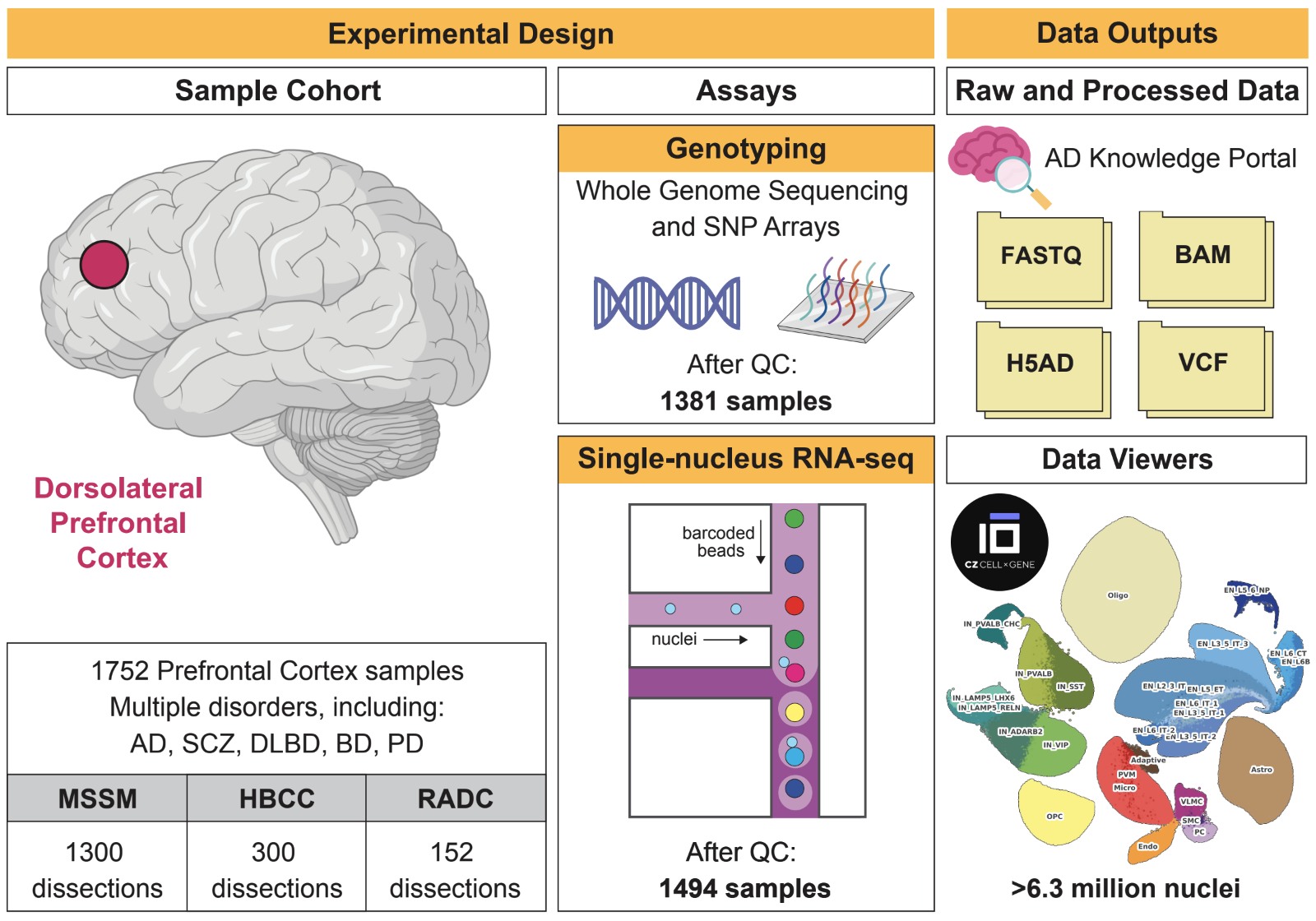
Neurodegenerative diseases and serious mental illnesses often exhibit overlapping characteristics, highlighting the potential for shared underlying mechanisms. To facilitate a deeper understanding of these diseases and pave the way for more effective treatments, we have generated a population-scale multi-omics dataset consisting of genotype and single-nucleus transcriptome data from the prefrontal cortex of frozen human brain specimens. Encompassing over 6.3 million nuclei from 1,494 donors, our dataset represents a diverse range of neurodegenerative and serious mental illnesses, including Alzheimer’s and Parkinson’s diseases, schizophrenia, bipolar disorder and diffuse Lewy body dementia, as well as neurotypical controls. Notably, 48% of all donors displayed neuropsychiatric symptoms, while over 21% received multiple diagnoses. The dataset presents an unprecedented opportunity to investigate both common and distinct molecular pathways spanning a range of brain-related disorders. We have performed stringent preprocessing and quality controls, ensuring the reliability and usability of the data. As a commitment to fostering collaborative research, we provide this valuable resource as an online repository, enabling widespread analyses across the scientific community.